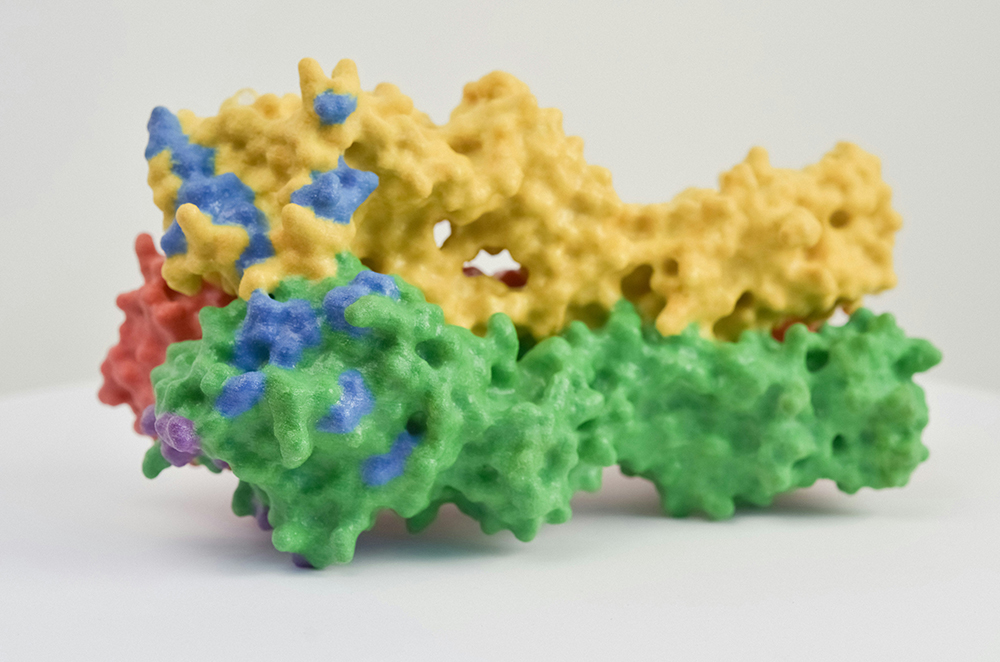
蛋白-配体互作分析(2D & 3D)
在完成蛋白和分子的对接后,我们会得到蛋白和分子的亲和力得分值,当然不同的软件,如Sybyl、GOLD、AutodockVina等,这些软件的得分取值范围存在存在很大的差异,这里我们就不进行详细的讨论了。在这里我想和大家分享的是关于对接的后续分析。
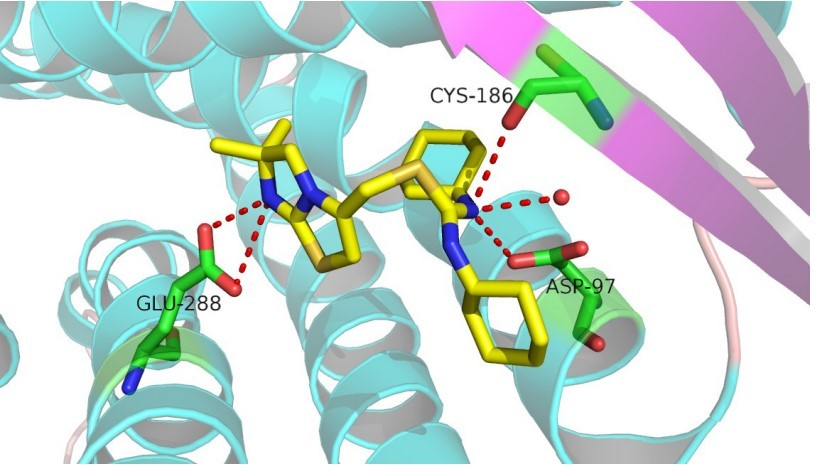
我们经常的论文中看到下面两种类型的图,他们分别表示蛋白和分子相互作用情况的3D和2D示意图,我们会看到分子通过氢键的作用里与蛋白上的氨基酸产生相互作用。这个就是我们今天要介绍的内容。
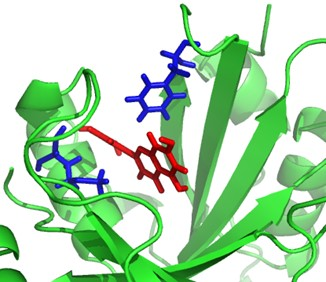
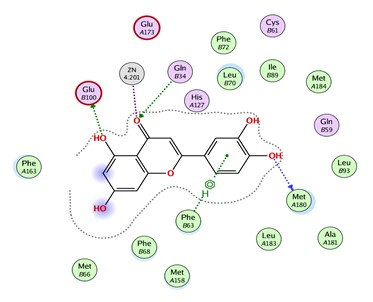
如何获得上述的结果?我们需要准备什么?请大家带着这两个问题开始今天的介绍。
1.蛋白分子互作分析-准备篇
在开始蛋白-分子的互作分析前,我们当然需要一个蛋白-分子结合文件图,即一个PDB文件。通常,在使用软件完成蛋白-分子对接后,我就能利用对应的软件去提取蛋白-分子结合的文件,即PDB文件。不同的软件有不同的方法,
以Autodock-Vina为例,在我的另外两个博文中,我已经介绍了如何使用Autodock-Vina进行对接,并提供了一个在windows上运行的案例。
1.1.案例:我们对接了PTGS2和Paeonol分子
PTGS2.pdb:原始的受体蛋白,包含了这个晶体自带的配体
PTGS2.pdbqt:受体蛋白,经过处理从PDB转化成PDBQT格式wend
Paeonol.pdbqt:小分子,我是利用Open Babel直接从MOL2格式转成PDBQT格式
Paeonol.zip:Windows下,利用PaDEL-ADV对接运行完成后,产生的对接结果压缩文档
result.csv:记录对接得分的csv文件
1.2.Paeonol.zip中内容:
Paeonol.out.pdbqt:对接运行记录文档
Paeonol.out_ligand_1.pdb:对接得分1中小分子pdb格式文件,小分子的空间结构发生了改变以便与PTGS2结合,同时其记录的分子结构的空间坐标与原始的分子文件不同,当前分子的空间坐标已经与PTGS2受体文件的空间相适应,以便于后期组装成蛋白-分子复合物。不同的得分2-9,分别对应不同的分子文件
Paeonol.out_ligand_1.pdb:对接得分1中小分子PDBQT格式文件,.....
Paeonol.pdb:原始的Paeonol小分子PDB格式文件
Paeonol.pdbqt:原始的Paeonol小分子PDBQT格式文件
1.3.文件列表的结构:
--PTGS2
---PTGS2.pdb
---PTGS2.pdbqt
---Paeonol.pdbqt
---Paeonol.zip
---result.csv
Paeonol.zip解压后得到如下对接的结果:
--Paeonol
---Paeonol.log.txt
---Paeonol.out.pdbqt
---Paeonol.out_ligand_1.pdb
---Paeonol.out_ligand_1.pdbqt
——————————————————————————————————————
---Paeonol.out_ligand_9.pdb
---Paeonol.out_ligand_9.pdbqt
---Paeonol.pdb
---Paeonol.pdbqt
1.4.获得蛋白-分子复合物
简单讲就是我们要得到一个PDB文件,这个文件中包含了蛋白,分子的空间坐标信息。
先看看我们有什么文件?
蛋白受体:PTGS2.pdbqt 或者 PTGS2.pdb
分子文件:Paeonol.out_ligand_1.pdb 或者 Paeonol.out_ligand_1.pdbqt,以及后续的2,3,...,9
我们有两个方法来出来达到我们的目的:1)借助PyMOL软件,组合蛋白和分子文件获得复合物文件;2)借助Linux的Shell命令来获得相应的复合物文件
1.4.1.PyMOL方法获得复合物
具体的操作过程如下:
利用PyMOL打开受体蛋白文件(PDB)
通过菜单栏打开分子的文件:"File" --- "Open..."
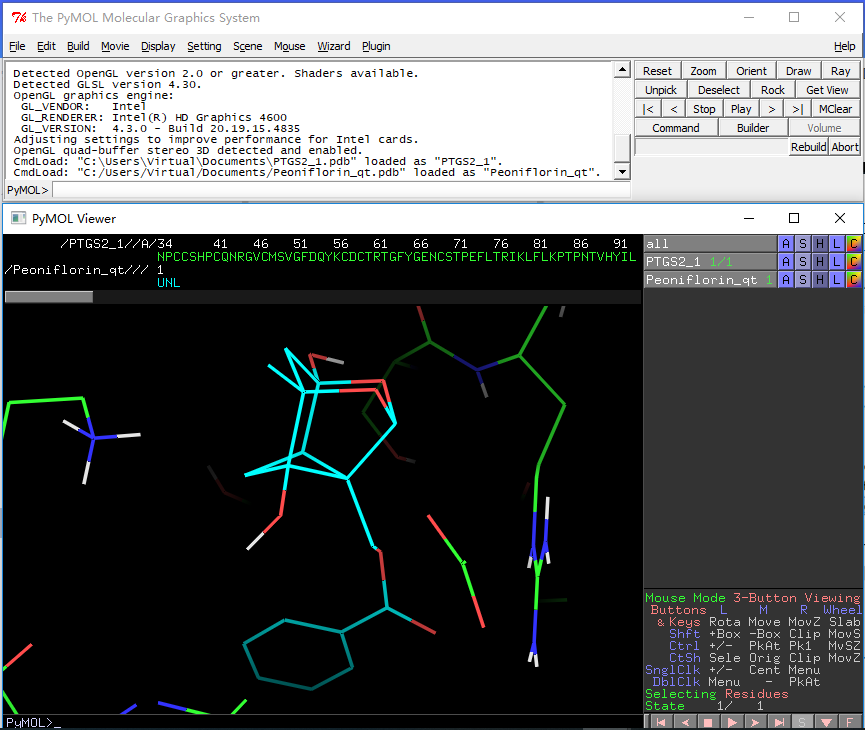
选择我们需要的蛋白和分子
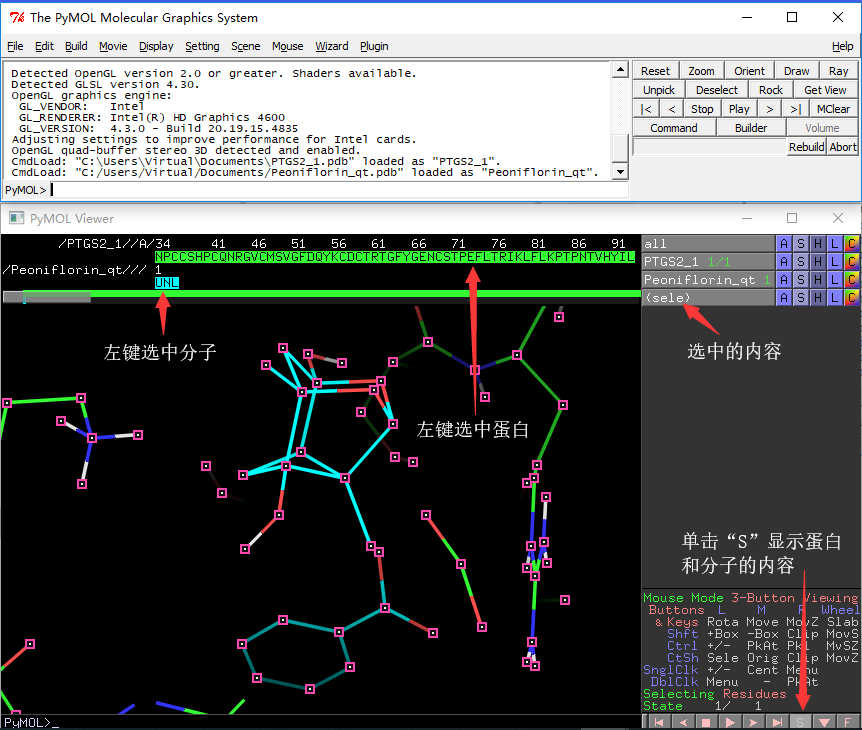
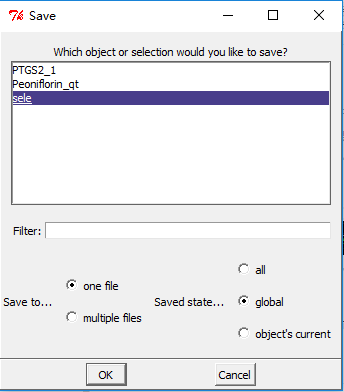
通过菜单栏保存我们的选中的内容:"File" --- "Save Molecule..." --- "sele" --- "ok" --- "保存"
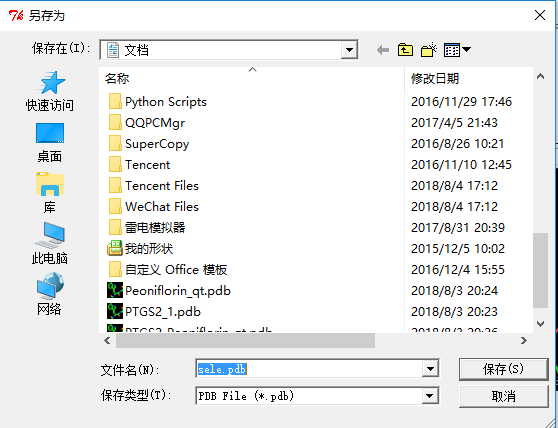
多啰嗦几句,目前我们就保存了蛋白-分子复合物的PDB文件,实际上,上述操作归结起来就是手动的去合并多个PDB文件的全部或者一部分到一个PDB文件中,所以大家如果有类似的操作需求,就可以参考上面的过程去操作获得一个目标PDB文件。
1.4.2.Shell命令获得复合物
PyMOL是常用的一个操作PDB文件的方法,同时Autodock也提供了相应方法来获得我们的目标复合物,具体大家可以参考下面的官网的方案:
下面我们就根据我们的需求具体实操一下,我们以PTGS2.pdbqt、Paeonol.out_ligand_1.pdb、Paeonol.out_ligand_1.pdbqt为例,将他们放在同一个目录下(实际上,可以不同目录,到时输入命令的时候需要指定详细的路径)
将PDBQT转化为PDB(Converting from PDBQT to PDB)
要将PDBQT格式转换为PDB格式,最简单的方法是删除PDBQT文件中的电荷(charge,Q)和原子类型(atom type,T)列,具体的可以使用Shell命令:
$ cut -c-66 PTGS2.pdbqt > PTGS2.pdb $ cut -c-66 Paeonol.out_ligand_1.pdbqt > Paeonol.out_ligand_1.pdb创0建复合物的PDB文件(Creating a PDB File of the Complex)
为了创建包含受体蛋白和单个小分子配体的复合物PDB文件,我们需要使用Unix的“cat”命令将蛋白文件和分子文件连接起来,然后使用带“grep -v”提取文件中除了带“END”符号的全部行。这个命令最终产生了一个带蛋白配体和分子的PDB文件,即我们需要的复合物。具体命令如下:
$ cat PTGS2.pdb Paeonol.out_ligand_1.pdb | grep -v '^END ' | grep -v '^END$' > PTGS2_Paeonol.pdb
上述步骤需要需要在Linux环境下,利用Unix的命令才能完成复合物PDB文件的合成。如果你是Linux用户,不用说一切顺利;如果你是Windows用户,目前有多个可以运行Linux终端的工具,比如:Cygwin,Window子系统等。这里我推荐MobaXterm,这个是一个既可以本地运行Linux终端,也可以远程连接服务器的强大Windows全能终端神器,具体信息大家可以自己去检索
到此,准备工作结束了,我们获得了一个复合物文件“PTGS2_Paeonol.pdb”。下面就可以蛋白-配体2D/3D互作分析图
2.蛋白分子互作分析-3D分析篇
做蛋白-小分子3D结合模式分析的工具还是挺多的,在这里给大家介绍的主要是两个工具:PyMOL和PLIP。这两个工具是我自己自己网上检索后,发现比较好的开源,免费的工具。当然很多商业的药物设计软件也是有这些功能的,但是就科学研究来说,大家的资金大部分情况下是并不充足的,因此这里只是推荐开源免费的工具。就个人感觉来说这两个工具也是用的最多的。
2.1.PyMOL - 蛋白-分子3D结合模式分析
蛋白-分子互作的3D分析,大部分是用开源软件PyMOL进行分析的,相关的内容,网上的教程也有很多。我以后有时间会分享出来,或者大家自行检索。
这里给大家推荐一个教程
http://440d8b07.wiz03.com/share/s/143oI73NWQdH2vUPFq3KhBcA2-0ZXv2ALAw12Q5aQm07-KEV 密码:g322
总的来说,大家可能吐槽是要获得后期下面这样的图需要进行好多步操作,并且从我目前实践看,目前得到的分析结果只有小分子氢键结合的氨基酸残基及其距离。可能还有好多分析结果,但是目前我还没发现,期待大家能进一步发现新的分析结果。
说了槽点,我们说说PyMOL的优点,实际上使用PyMOL可以很好的定制我们最终呈现的效果图,在颜色、布局等等上都可以自己调节以便获得让清晰的结果。此外PyMOL还有很多插件可以辅助大家进行分析,实话说,利用好了插件,你就能得到更好的分析结果。所以有时间,大家可以去检索一下PyMOL有哪些高效、好用的插件来辅助研究分析。
2.2.PLIP- 全自动 protein–ligand interaction profiler
在你使用PyMOL后,在你遇见PLIP后,你会爱上这个工具的。这个工具可以轻松快速地识别蛋白质与其配体之间的非共价相互作用。接着看,你就会知道什么叫轻松快速?
简介:PILP(Protein-Ligand Interaction Profiler)是一个在线的蛋白配体非共价联系的分析工具,其简单易用,同时提供Github版本利于学习其设计模式。PILP可以直接使用四字符的PDB code或者使用自己的pdb结构文件。之后分析复合物,结果将会展示所有可测定的在原子水平的非共价联系(氢键,水桥,盐桥,卤键,疏水联系,π-堆积,π-离子联系,金属复合物)。同时结果可以下载文本结果,机器可读的XML格式以及PyMOL可视化文件(pse),同时网站上也有JSmol应用的3D联系图像。需要再所一下的是PILP也提供了脚本处理工具,可以得到Web版的结果,具体可以在其Github中查看。
网址:https://projects.biotec.tu-dresden.de/plip-web/plip/
Github地址:https://github.com/ssalentin/plip
实操:我们以PLIP的Web应用为例介绍如何使用这个工具,我们先来看看它的界面:
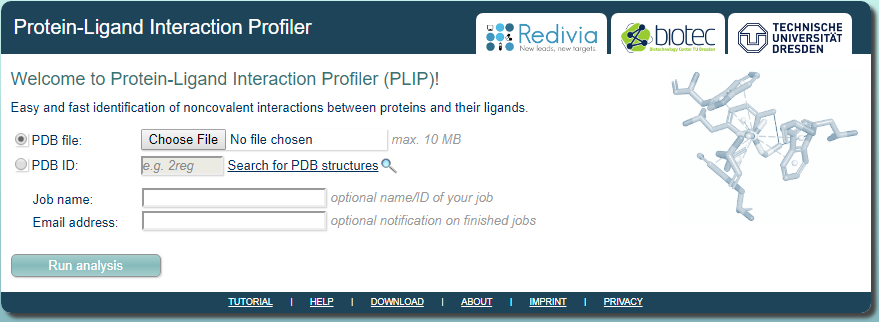
通过“Chosse File”提交我们的蛋白-分子复合物PDB文件,并开始运行。这里我们选择准备好的 “PTGS2_Peoniflorin_qt.pdb” 文件。等待一会后,我们会得到一下的结果:
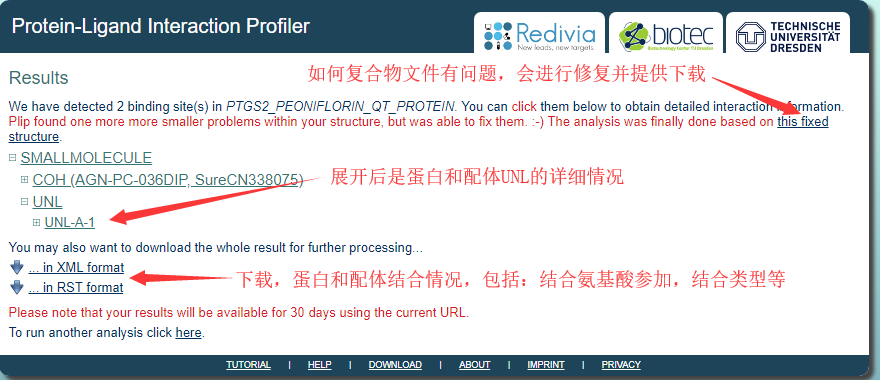
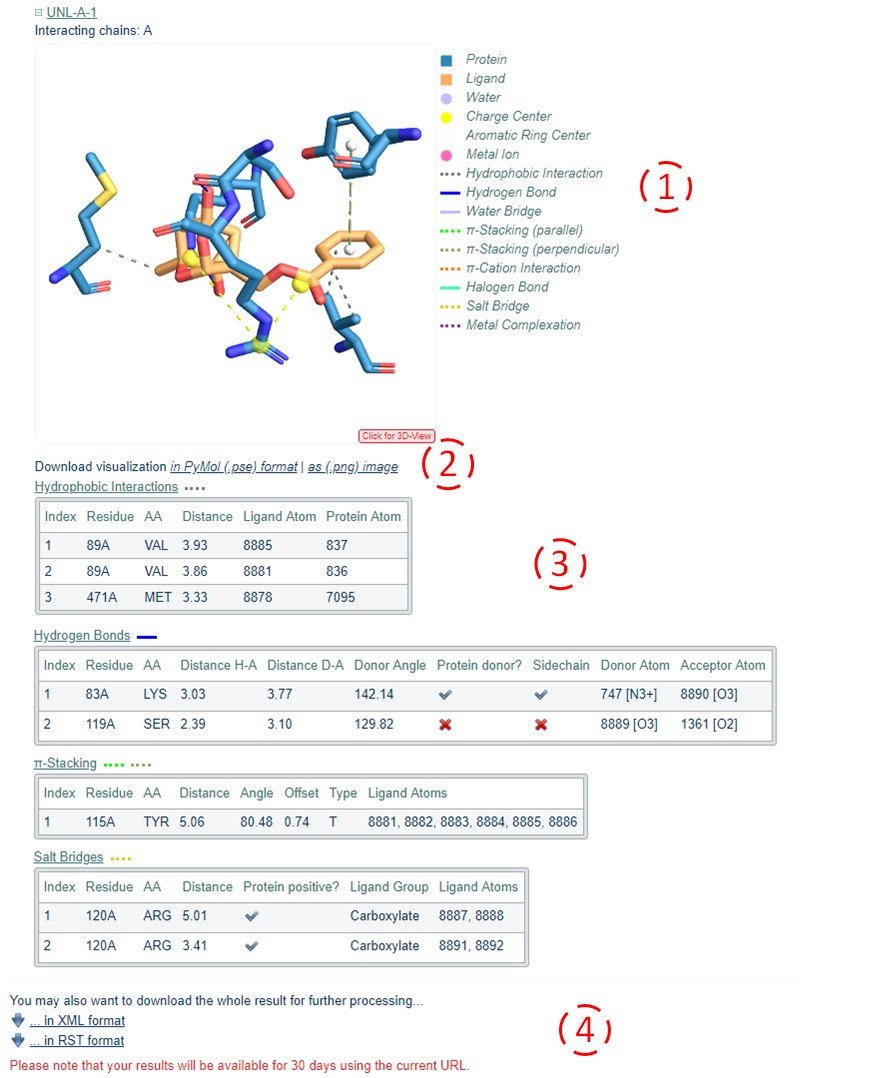
在上面这些内容中,我们需要仔细关注的是“UNL-A-1”展开之后的内容,下面就进行一个详细的介绍:
内容1:对于结构中的每个蛋白质 - 配体复合物,PLIP显示了渲染的相互作用图。 配体显示为橙色,蛋白质残留物为蓝色。 非共价相互作用由虚线或实线表示,如右侧图例中所示。如果要进一步观察交互式三维可视化图,可以单击预览图像以打开JSMol小程序。通过这个小程序可以观察相互作用图的不同视角。
内容2:这里提供了两个下载选项:1)“as (.png) image”可以高分辨率下载内容1中的渲染图像;2)“ in PyMol (.pse) format”可以下载用于渲染图像的PyMOL会话文件,这个文件可以被PyMOL 1.7.x及后续版本打开。通过这个文件,我们可以继续观察相互作用,选择我们需要的配色方案,显示的文字内容等等个性化修改。实际上回想PyMOL单独处理蛋白-分子复合物PDB文件复杂过程,是不是能看到这个工具的便捷性和易用性。
内容3:对于每一种交互类型,列表会展示出原子级的详细信息。 表格中的每个条目包含有关蛋白氨基酸残基、参与互作的配体的原子和相互作用几何形状(例如相互作用原子的距离)的详细信息。 根据不同的互作类型,可获得额外的信息,例如:用于π-stacking(π-堆叠)的芳环的偏移。此外,我们可以通过鼠标悬停在条目上显示每个属性的详细说明。 而且所有原子和残差编号都与输入PDB文件中的编号一致。这里,再说一句,PLIP对于如何判断蛋白-分子之间的互作类型是根据一套规则来的,详细的信息,大家可以访问PLIP的帮助中心查看(https://projects.biotec.tu-dresden.de/plip-web/plip/help)
内容4:为了后续的处理或检查,您可以下载两种类型的数据 :XML和RST格式的文件。这些文件中包含了上述分析中的所有配体的交互数据。 并将结果页面上看到的表格以人类可读格式呈现。对于新手,个人推荐RST格式的文件。如果你对xml文件很熟悉,可以下载XML格式的文件去处理
到此,我们基本就利用PLIP完成了蛋白-分子复合物3D互作的分析。而关于PLIP如何进行脚本运行,批量分析,这里就不在多讲了,有兴趣的话,大家可以去PLIP的Github的地址学习。此外对于这个工具的学习和认识,大家还可以参考其官网的帮助和快速入门:
3.蛋白分子互作分析-2D分析篇
做蛋白-小分子2D结合模式分析的工具还是挺多的,像MOE的等软件就能给出很好的图像,可惜MOE是一个商业软件,这里我们还是介绍几款开源免费的软件:LigPlot+、PoseView 和 PoseView Web版(http://poseview.zbh.uni-hamburg.de/)。需要注意的这些软件对于科学研究者是免费的,只要我们通过研究者的单位邮箱去申请“Academic licence”就可以免费运行这些软件,注意不要用像gmail,163,qq等邮箱去申请。接下来我们详细的介绍各个软件:
3.1.LigPlot+ - 经典配体与蛋白二维相互作用作图软件
LigPlot+是LigPlot的GUI界面化软件,是一款经典的配体与蛋白二维相互作用作图软件,可展示配体分子与受体蛋白之间的氢键、疏水等相互作用。同时还支持PyMOL和Rasmol的三维图像。
下载地址:https://www.ebi.ac.uk/thornton-srv/software/LigPlus/
操作简介:
申请“Academic licence”
申请地址:https://www.ebi.ac.uk/thornton-srv/software/LigPlus/applicence.html
填写真实的信息和单位信息,尤其注意邮箱的填写,和你的单位保持一致。就国内来说,大部分高校的邮箱都是可以的,他们的邮箱都带有edu.cn。比如西北农林科技大学:@nwafu.edu.cn
下载软件
下载地址:https://www.ebi.ac.uk/thornton-srv/software/LigPlus/download.html
在申请“Academic licence”后,一般在1-2天左右,EBI会给你填写的邮件发送一份邮件,里面包含了你的E-mail和Password,这些信息将用来下载软件。
邮件信息:
下载地址:
安装软件
实际上下载的是一个3兆多的压缩包“LigPlus.zip”,不多说,直接解压到一个纯英文路径的目录下,我们可以看到,这个目录下包含两个文件和一个目录:
其中运行的主要软件是“LigPlus.jar”,看到这个我们就知道要运行它,必须在系统中安装java的运行环境,大部分的电脑都应该安装了java,所以不用担心,如果没有,那就去下载安装吧(https://www.java.com/zh_CN/)。
1)左键双击运行“LigPlus.jar”后,我们就能看到如下的界面,因为是第一次运行,所以需要设置一些基本的设置
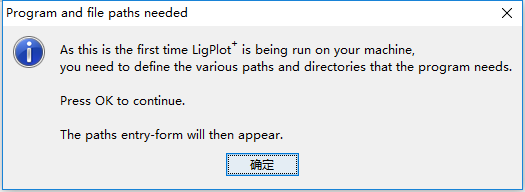
2)根据自己的喜好设置我们喜欢的路径:如果你修改了你的路径,请手动的新建这些目录,因为LigPlus不会自动创建这些目录
3)设置成功,开启LigPlus+软件
LigPlus+开始运行分析
首先从菜单栏打开我们的蛋白-分子复合物PDB文件:“File” --- "Open >" --- "PDB file" --- "Browse" --- "选择文件" -- --“open” --- “选择配体” --- “Run”
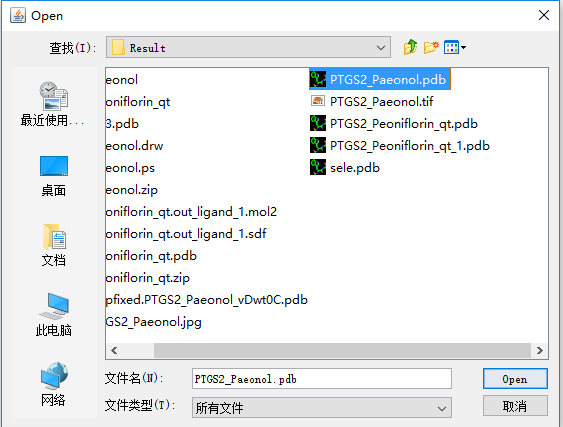
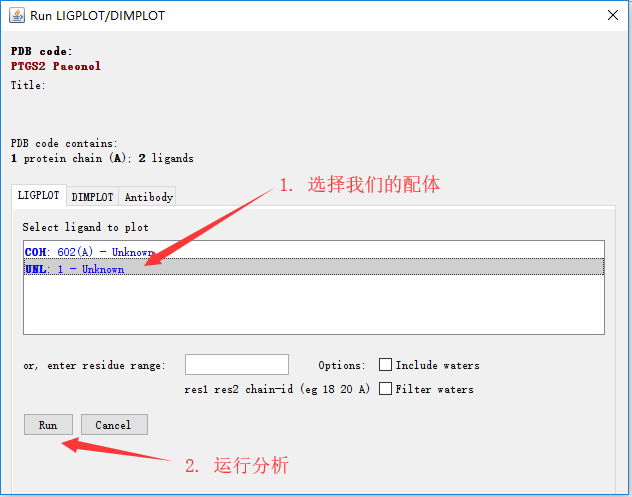
查看结果并保存
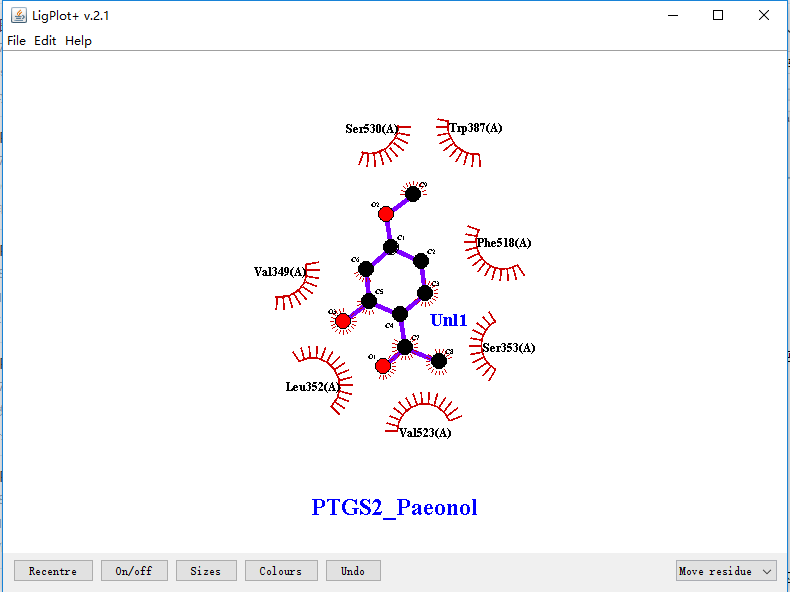
可以看到下面的结果,在这里我们可以通过鼠标左键进行氨基酸或者配体的拖动,或者鼠标右键修改上面的文字。剩下的健的粗细,颜色等等修改,大家可以自行去探索。
当你觉的当前的展示形式符合你的要求了,你就可以保存结果了,目前有两种保存格式:
drw格式:“File” --- "Save" --- "选择保存路径及文件名" --- “Save”
ps格式:“File” --- "Wirte PS file" --- "选择保存路径及文件名" --- “Save” --- “选择保存参数:默认即可” --- “OK” --- “确定”
就我个人而言,我喜欢保存ps格式的文件,这样我后续就可以通过Adobe的Photoshop软件或illustrator软件去编辑作为最后的展示结果。
闲话
至此我们就基本完成了基于LigPlot分析的二维蛋白-分子互作。我们想说,这个软件还不是百分百顺利运行的,有时候我们会遇到一些问题,这些问题请大家Google搜索去解决这些问题。此外,大家也可以关注以下几个网址继续了解LigPlot,也许有些问题就能在这里找到:
LigPlot+依赖的LigPlot程序地址:https://www.ebi.ac.uk/thornton-srv/software/LIGPLOT/
LigPlot+ v.1.4的教程:https://www.ebi.ac.uk/thornton-srv/software/LigPlus/manual/manual.html
LigPlot+ v.2.1的教程:https://www.ebi.ac.uk/thornton-srv/software/LigPlus/manual2/manual.html
3.2.PoseView Web版
PoseView Web版是是一个基于PoseView的蛋白-配体二维互作分析的在线工具。它的使用也很简单,我们就来看看吧:
登录界面
登录地址:http://poseview.zbh.uni-hamburg.de
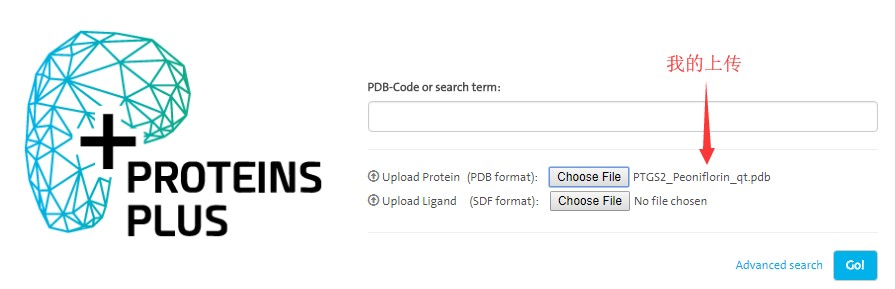
这个就是PoseView Web版。我们只需要上传我们的复合物的PDB文件,这里我们选择“PTGS2_Peoniflorin_qt.pdb”,此外我们也可以将蛋白和分子分开上传。
命令:蛋白部分-“Choose File” --- "选择复合物PDB文件" --- “Gol”
分析界面
这就是PoseView Web版的分析界面,主要由三个板块组成:
板块1:PDB文件渲染图,可以旋转、放大观察
板块2:配体的信息,用于后续功能分析的配体选择
板块3:分析功能,我们可以看到,这里有很多的分析功能,目前我们需要看的是“PoseView 2D interaction diagrams”
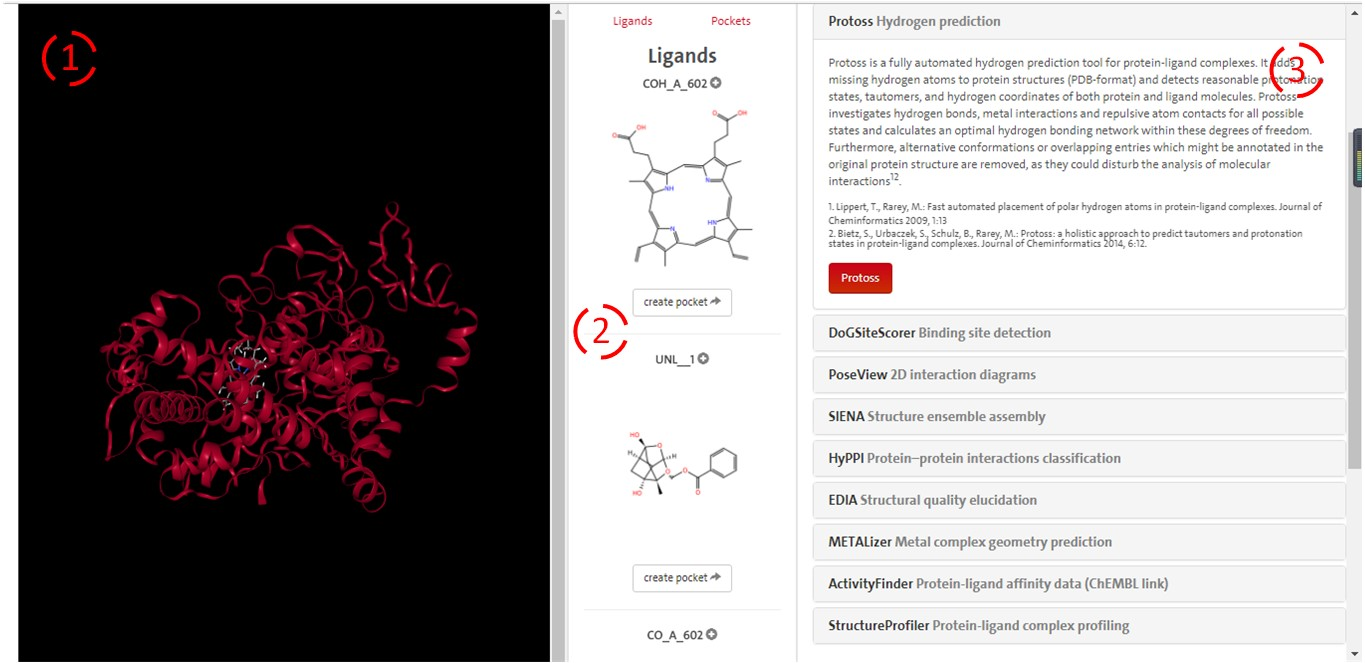
“PoseView 2D interaction diagrams”分析:
我们先开启“PoseView 2D interaction diagrams”工具,并选择配体,然后开始运行分析,如下所示。
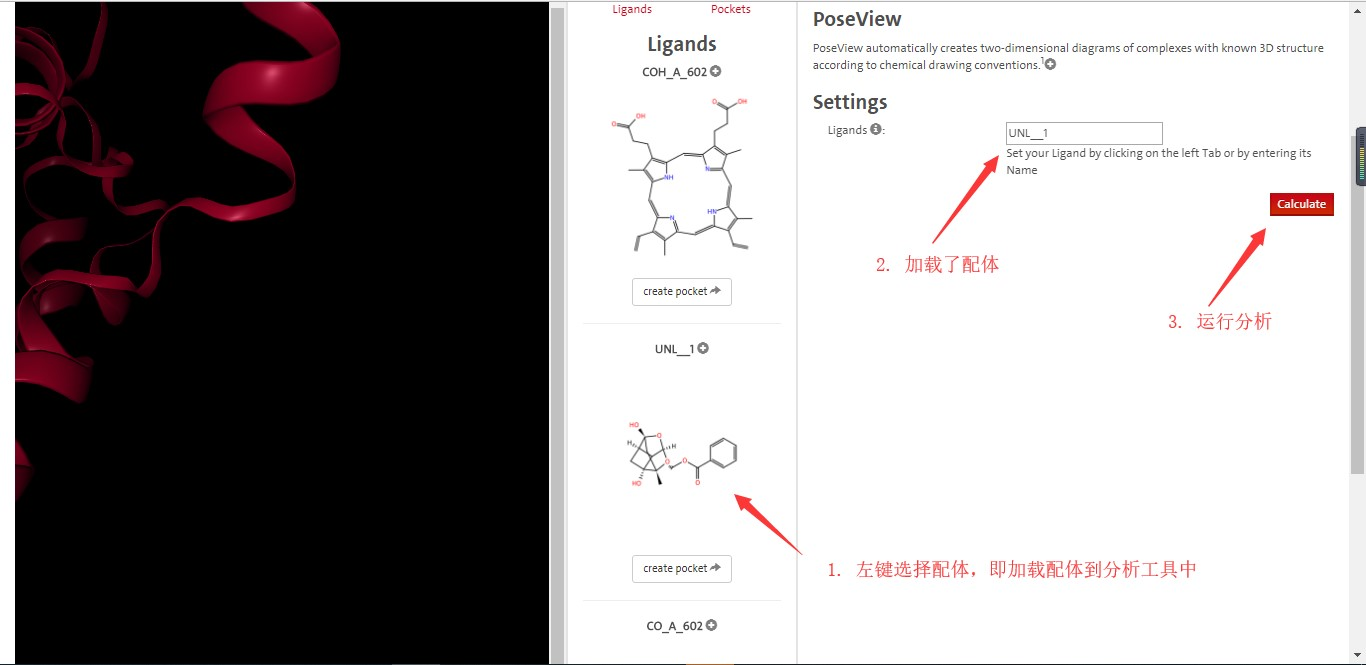
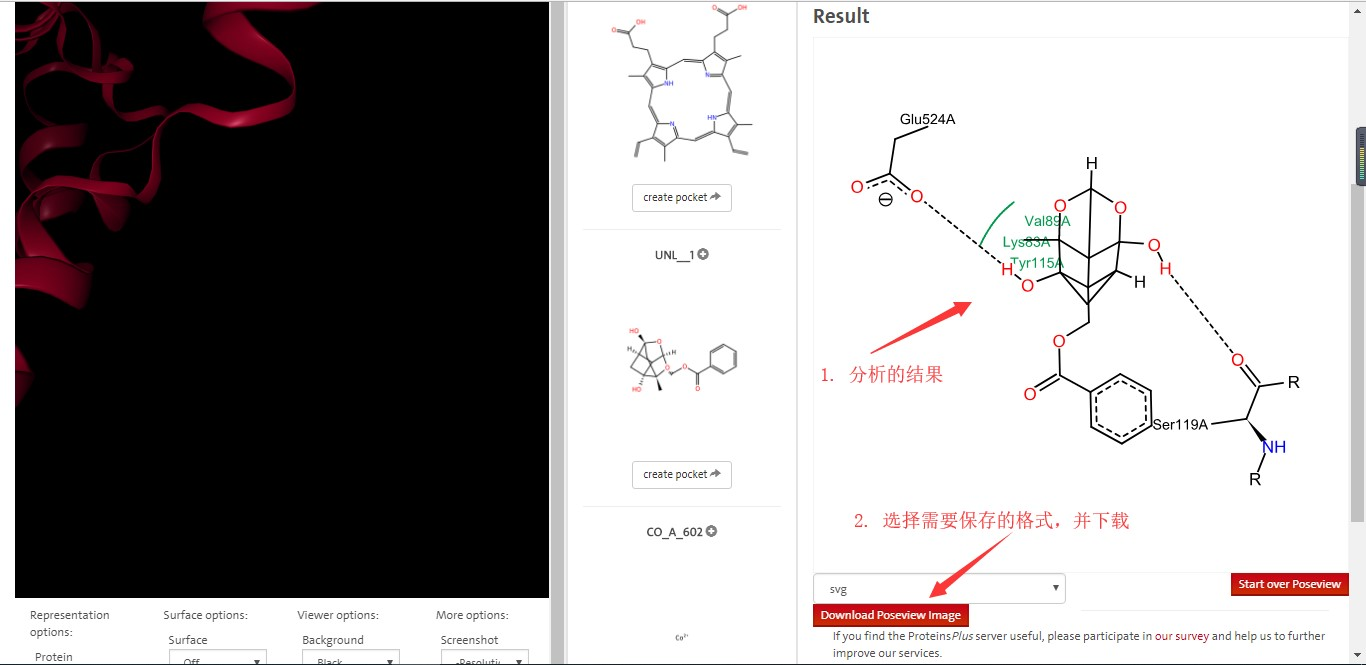
保存分析结果
如图所示,途中显示的就是蛋白-分子的二维互作结果,两个氨基酸与小分子具有氢键作用。
后续我们就要保存这个结果作为我们论文的一部分,这里提供了三种格式的下载:svg,pdf,png。就我个人而言,我们喜欢保存pdf,这样可以后续接着进行图片的后续处理,如决定保存的图片的分辨率大小等。
闲话
实际上这个网页工具还有很多有用的功能,大家可以继续去体验一下,操作过程都差不多。这里需要提一下,这个网页是基于PoseView软件的,我们是否可以直接用这个工具呢?下面我们就来介绍PoseView这个软件。
3.3.PoseView软件
PoseView强大的蛋白-分子2D互作分析的软件,它可以在2D中可视化蛋白质 - 配体复合物,自动生成出版品质的图形。而且这个工具也我们常用的PDB数据库(https://www.rcsb.org/)中蛋白-配体互作2D可视化的工具。而且它给我的感觉是最接近MOE软件蛋白-配体2D互作分析结果的一款软件。当然PoseView是一个商业软件,但是它对于学术研究是免费的,需要我们去申请相应的licence,现在让我们来看看它具体是如何操作的。
3.3.1.软件使用权申请
在使用PoseView软件前,我们先需要申请其“Licence”,具体的申请地址是:https://www.biosolveit.de/PoseView/
我们首先登录PoseView的官网, 找到“request a free license for academic research”,左键单击后,会出现一个邮件会话界面:我们要向“license@biosolveit.de”发送一份主题是“free PoseView license for academic research”的邮件,填写Licence申请。下面以我和对方的交流说一下“Licence”申请的整个过程
PoseView的官网

第一次发送申请邮件
每一次给对方发送一份邮件,对方都会自动发送一份受理的邮件,这个不用理会。
第一次收到回复邮件
实际上就是让我准备一个类似合同的文件,扫描后发送给对方
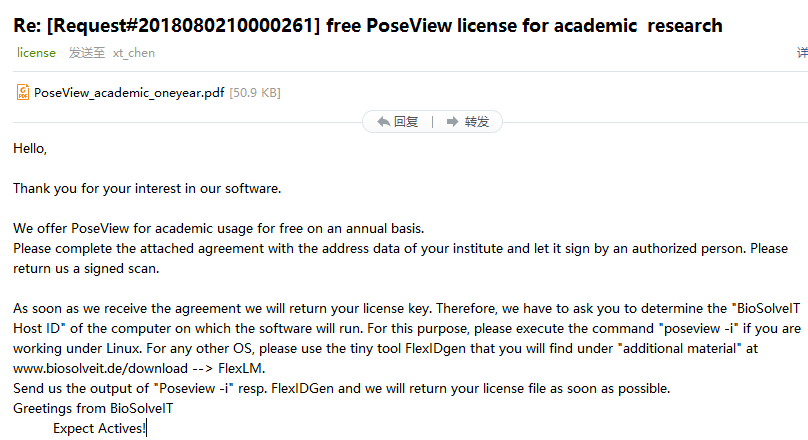
合同文件准备
填写邮件的中的PDB文件,其中要注意是电脑"BioSolveIT Host ID"的准备,这是因为PoseView学术免费版针对的是一台电脑一个软件,因此"BioSolveIT Host ID"就相当于我们电脑的身份证。我们参考邮件中提示就能完成。
我的要安装的电脑Windows系统,下面我就以Window为例说来说如何进行填写:
1)"BioSolveIT Host ID"计算软件下载
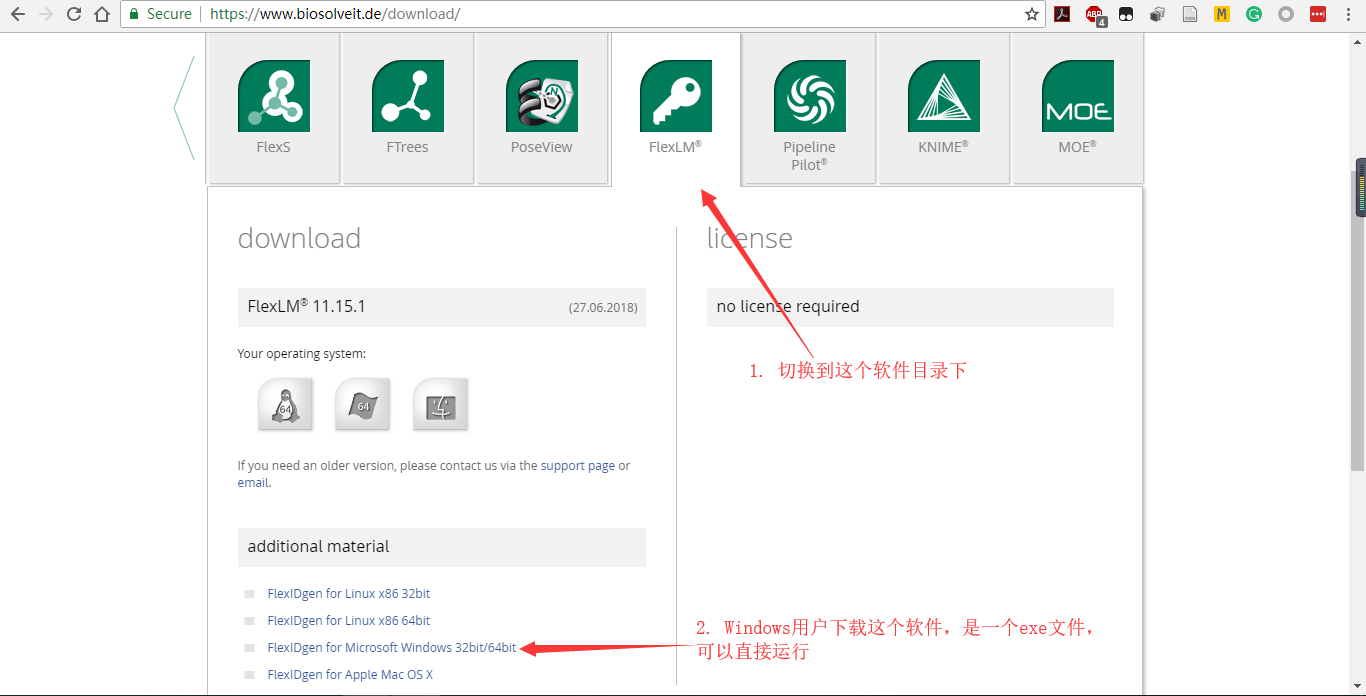
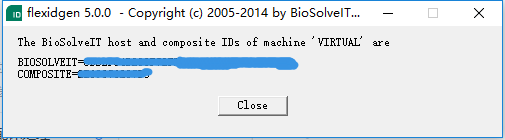
2)本地运行“flexidgen-5-Win32.exe”获得对应的"BioSolveIT Host ID",保存对应的值
3)填写合同PDB文件





回复邮件
将填写好的PDB打印扫描一份,并作为附件,回复给对方,最好在邮件正文中加上由“flexidgen-5-Win32.exe”获得的"BioSolveIT Host ID"。当然我刚开始的时候没有填写,但是后来对方也发邮件向我要了。
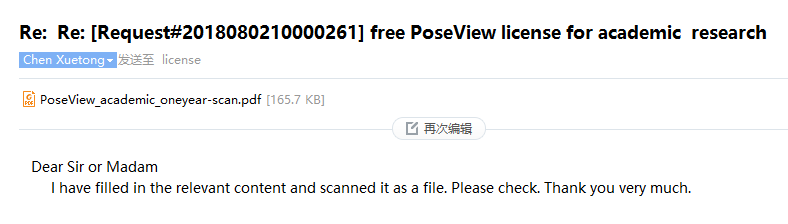
我们收到对方的回复邮件
获得了“License”,成功申请。
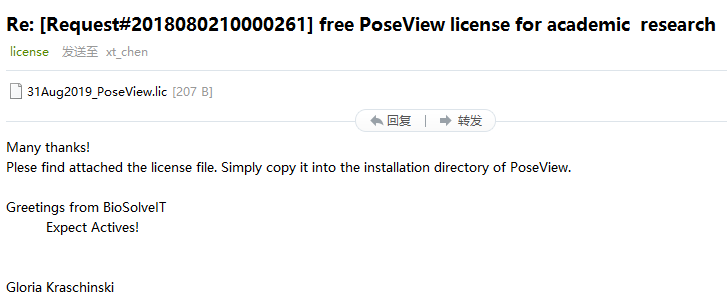
3.3.2.安装PoseView软件
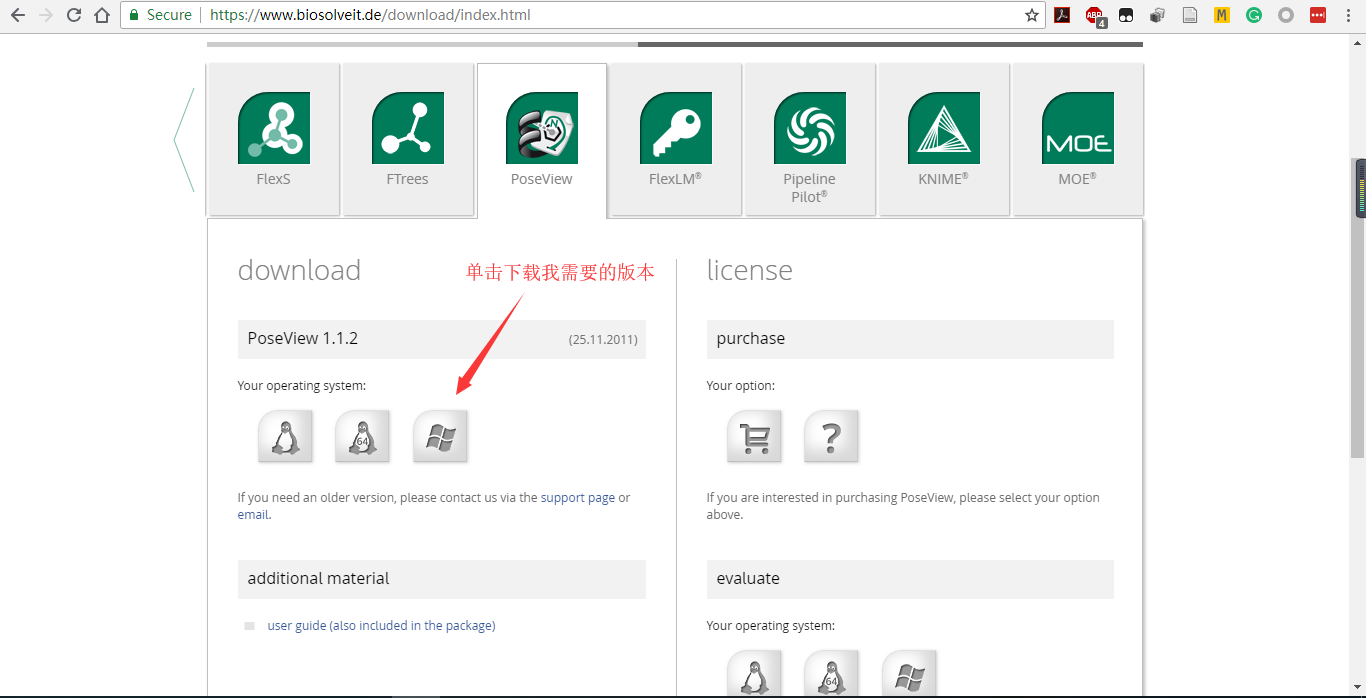
软件下载地址:https://www.biosolveit.de/download/index.html
在获得权限后,我们下载PoseView软件(poseview-1.1.2-Win32.exe)并安装。安装过程我就不说了。
下载安装软件
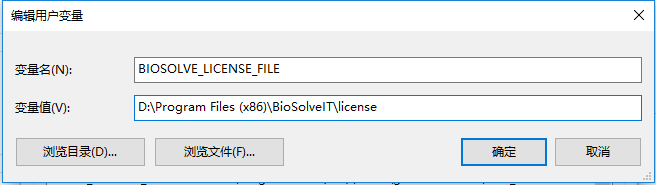
设置License的环境变量
将邮件回复中的PoseView的License文件保存在安装目录的license目录下(D:\Program Files (x86)\BioSolveIT\license)。并设置环境变量:
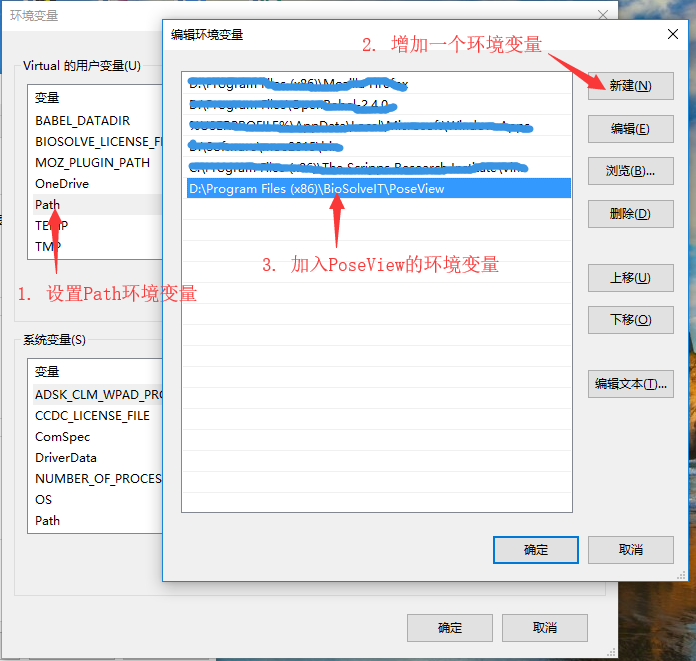
设置软件的的环境变量
将安装的PoseView的路径(D:\Program Files (x86)\BioSolveIT\PoseView)加入,“Path”环境变量中,以便在CMD等终端环境调用PoseView软件
3.3.3.运行PoseView软件
我们打开软件快捷方式,得到如下界面:
分析复合物蛋白-分子2D互作情况
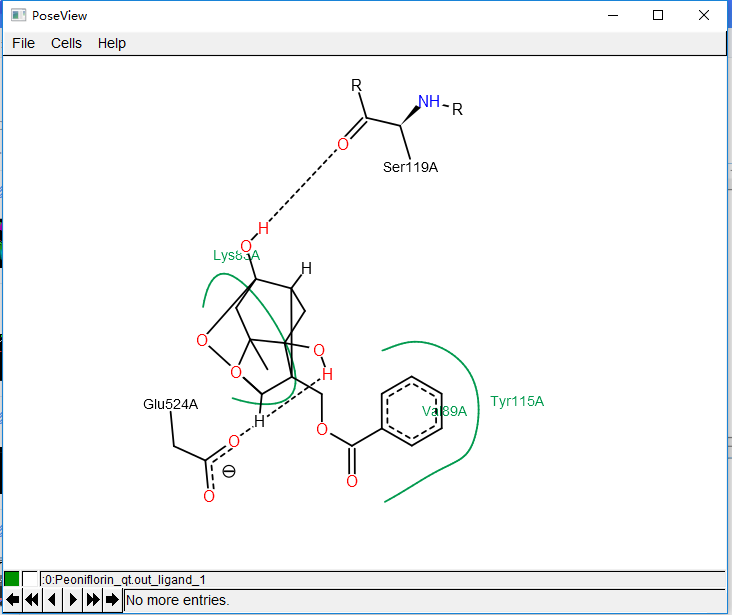
我们打开软件,导入复合物PDB文件,查看蛋白-分子二维的互作情况。
命令:“File” --- “Open Files” --- “Ligand and Protein File” --- "分别导入配体小分子和蛋白"
3.3.4.保存PoseView分析结果
当前的软件在图形界面上没有保存结果图片的功能,但是我们可以通过CMD终端上的命令保存我们的分析结果,我们首先打开CMD界面,然后切换到有蛋白和配体文件的目录,输入以下命令:
# cmd命令界面
C:\Users\Virtual\Documents> poseview.exe -l Peoniflorin_qt.out_ligand_1.sdf -p PTGS2_1.pdb -o PTGS2_Peoniflorin_qt.png -s 1000 10000通过这一步我们就能得到我们需要分析结果,图片格式是png,具体的参数,我们可以调用出来,大家看看:
# cmd命令界面
C:\Users\Virtual\Documents> poseview.exe -h
Qt: Untested Windows version 6.2 detected!
______________________________________________________________________________
___ __ ___
| _ \___ ___ __\ \ / (_)_____ __ __
| _/ _ (_-</ -_) V /| / -_) V V /
|_| \___/__/\___|\_/ |_\___|\_/\_/
ZBH - Center for Automatic generation of 2D complex diagrams
Bioinformatics
University of Hamburg Version: 1.1.2 (25.11.11)
Bundesstrasse 43
20146 Hamburg
Germany Authors: Katrin Stierand, Matthias Rarey
BioSolveIT GmbH Copyright: ZBH, University of Hamburg, Germany
An der Ziegelei 79 BioSolveIT GmbH, Sankt Augustin, Germany
53757 St. Augustin Contact: poseview@zbh.uni-hamburg.de
Germany www.zbh.uni-hamburg.de/poseview
______________________________________________________________________________
For information about additional contributors and copyright notes
please consult the user guide or type 'help about'.
>>> SYNOPSIS:
poseview [OPTIONS]
>>> OPTIONS:
-c <configfile> : Configuration file (xml format)
-f <filename> : Read input data from complex file (.mol2)
-h : Printing of this help text
-i : Generate processor id for this machine
-l <ligfile> : The ligand is read from <ligfile>.
Default format is .mol2
-m <file> : List of ligand and protein file names.
Format: lp <ligfilename> <protfilename>
-o <outfile> : Prints the diagram to <outfile>
possible file formats: .png, .pdf, .ps, .fig, .svg
-p <protfile> : The protein is read from <protfile>. Default
format is .rdf, also accepted are .pdb, .fxx and .mol2
-s <w> <h> : Size of the .png or .fig output file.
png: 100 <= w|h <= 1000, fig 1000 <= w|h <= 50000
-t <text> : Info text printed in output file.
-v : Prints the version number and compilation
information.
-w <filename> : Write complex file from data (.mol2)3.3.5.其他
对于PoseView的了解还可以通过以下地址了解其结果的含义等等:
- 感谢你赐予我前进的力量
-
 微信
微信  支付宝
支付宝
