Autodock - 分子对接简介及操作
说明:本教程主要的内容来自Yunwen Tao、Xianlong Wang、Yunfeng Jiang、Bin He等人所做的教程,感谢这些作者的贡献
1. 分子对接的生物学背景
分子对接的最初思想起源于 Fisher.E 提出的酶与底物相互作用的“锁和钥匙模型",它指的是底物和酶(也就是分子对接中的配体和受体)之间通过几何匹配和能量匹配而相互识别的过程,也就是说分子之间的相互作用不仅是指参与对接两个分子在空间构象上的相互匹配,还要满足能量的匹配。即要求配体和受体间需要存在空间结构、氢键作用、静电作疏水作用等方面的互补匹配,这也就是所谓的互补匹配原则。根据对接分子对接配体、受体的体系大小和对体系简化程度的不同,分子对接方法可以分为三类:
刚性对接:刚性对接方法在计算过程中,参与对接的分子构像不发生变化,仅改变分子的空间位置与姿态,刚性对接方法的简化程度最高,计算量相对较小,适合于处理大分子之间的对接。
半柔性对接:半柔性对接方法允许对接过程中小分子构像发生一定程度的变化,但通常会固定大分子的构像,另外小分子构像的调整也可能受到一定程度的限制,如固定某些非关键部位的键长、键角等,半柔性对接方法兼顾计算量与模型的预测能力,是应用比较广泛的对接方法之一。之后介绍的 Autodock 系列对接软件使用的便是半柔性对接的方法。
柔性对接:柔性对接方法在对接过程中允许研究体系的构像发生自由变化,由于变量随着体系的原子数呈几何级数增长,因此柔性对接方法的计算量非常大,消耗计算机时很多,适合精确考察分子间识别情况。
但是幸运的是,目前已经存在了各种各样的分子对接软件如 Dock,Autodock,FlexX 和Affinity 等来实现我们的对接过程。而我们用户只需要做简单的准备工作和后续的分析工作即可。
2. 分子对接的主要流程和软件准备
2.1. 分子对接流程:
受体、配体分子坐标文件准备:
受体分子(通常是蛋白质)去除溶剂分子、杂分子,加极性氢,加电荷
从头绘制配体分子,加极性氢,加电荷,设置分子柔性等
格子参数的准备:格子(Grid)就是分子对接中用户指定的受体和配体可能发生相互作用的一个长方体区域,我们要在进行分子对接开始之前指定这个区域的位置和大小
分子对接:准备好的分子坐标文件和格子参数文件输入对接程序,运行后给出配体分子对接的构象,分析预测能量
结果分析:配体分子相似构象聚类分析、配体分子构象可视化分析、配体分子同受体分子相互作用可视化分析、AutoGrid 计算的亲和势能等
2.2. 软件准备:
我们所要使用的分子对接软件主要是 Autodock 系列对接软件中的 Autodock Vina,它是继 Scripps 实验室的 Autodock 4 对接程序之后由 Oleg Trott 等人推出的使用新的打分函数、预测准确率更高并支持并行化的更高效易用的对接程序。它的使用方法和 Autodock 4 类似,只是少了调用 AutoGrid 程序(原 Autodock 4 中需要用到)这一步。此外,在获得配体分子坐标文件时,可以采用三条条不同的途径:
从NCBI PubCompound数据库下载分子文件,保存为sdf后缀的文件
使用 Gaussview 从头画出配体的空间结构模型保存为 mol2 文件,稍微复杂的分子在画完后需要做一下量子化学水平的结构优化
如果配体十分复杂,可以先使用 ChemDraw 或 ChemBioDraw 画出配体结构的平面图,保存成 cdx 后缀的文件,然后使用 OpenBabel 转换成 mol2 文件

在准备输入给对接程序的分子坐标和格子参数时,我们需要使用的是 Autodock Tools, 它是和 Autodock 4 以及 Autodock Vina 配套使用的有用户图形界面的一个程序,同时在分析对接结果时,我们也会用到它。
更重要的是,我们需要一个 Linux 环境,来运行最后虚拟筛选部分涉及到的 Shell 脚本。
3. 分子对接过程与结果分析(以单个配体为例)
3.1. Autodock Vina 预处理
在本教程中,我们研究的体系是依赖 NAD+的 蛋白脱乙酰酶 Sirtuin 3 及其抑制剂的相互作用情况。Jeremy S. Disch1 等人已在实验室中筛选出了几种具有高效抑制功能的小分子化合物,并且将其中的 3 种抑制剂进行了与 Sirtuin 3 蛋白的共结晶并解析了 其晶体结构存放于PDB 数据库中。
从 Jeremy S. Disch 等人的工作中,我们了解到,Sirtuin 3 起催化功能时,NAD+辅酶与底物会结合到底物结合槽部位,而抑制剂结合时,NAD+将不再结合,仅有抑制剂结合到底物结合部位附近。考虑到 Sirt 3 结合不同的抑制剂时,蛋白的构象差异远小于结合底物时的情况,故我们以 结合了抑制剂的结构(PDB:4JSR)出发,开始我们的研究。
4JSR 中除了 Sirt 3 蛋白和抑制剂 1NQ 之外,还存在一个金属 Zn 离子和很多水分子的氧原子,但是我们期望探究其他抑制剂和 Sirt 3 蛋白相互作用的情况,所以要把抑制剂 1NQ 替换成我们预定的抑制剂(文献中的第 17 号抑制剂)。(Disch JS et al.Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3.J Med Chem. 2013 May 9;56(9):3666-79. )
基于上述信息,在进行Autodock对接前,我们需要对分子和蛋白进行处理,即对接体系(受体、配体)的确定与结构预处理。
3.1.1. 受体分子的预处理
打开 Autodock Tools 界面,可以看到如图的界面:
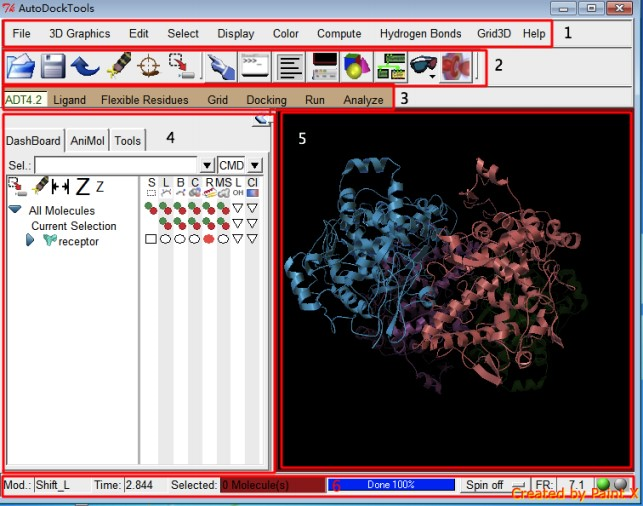
Figure 1: Autodock Tools 界面:1 PMV 菜单; 2 PMV 工具栏; 3 ADT 菜单; 4 仪表板窗口部件; 5 分子显示窗口; 6 信息栏
进行如下操作:
1)点击进入 File → Read Molecule 选择“4JSR.pdb”,打开受体分子,选择 Ribbon 类型的representation 表现方式,并按链(by chain)着色,可以看到只有一个亚基的存在。
2)点击 Select → Select From String 打开 Select From String 对话框,在 Residue 框中输入先存抑制剂的残基名称 1NQ*,点击 Add ,再点击 Dismiss 按钮关闭,新出现的黄色的部分就是自带的抑制剂 1NQ。
3)点击 Grid →Grid Box →滑动 Center Grid Box 的三个坐标滚柱,移动格子,并通过上面的三个滚柱调节格子的大小将选择的黄色抑制剂完全包围。最终选择的中心坐标为:20.031、46.672、-1.889,格子大小为 645850,记下这些数值。点击关闭 Grid Options 框。
4)点击 Select → Select From String 打开 Select From String 对话框,在 Residue 框中输入HOH,1NQ,在 Atom 框中输入*(通配符,表示所有原子),5)点击 Add 选中所有杂分子(溶剂分子和抑制剂分子)。最后,点击 Dismiss 按钮关闭 Select From String 对话框,可以看到水分子和抑制剂都被选取了。
6)点击 Edit → Delete → Delete Selected Atoms 删除已选择的分子,点击弹出的警告窗口上的 CONTINUE 按钮删除。
7)点击 Edit → Hydrogens → Add 为受体分子加 H(X 射线衍射无法获 H 的坐标数据。注意:加极性氢(连接在具有电负性的原子如 N、O、S 等上氢原子),并合并所有的非极性氢(连接在碳原子上的氢原子)点击 Edit → Hydrogens →Merge Non-Polar)。
8)点击 Edit → Charge → Add Kollman Charge 为受体分子加电荷。
9)点击 Grid → Macromolecule → Choose,选择后保存修改过的受体分子,这里保存为recptor.pdbqt
3.1.2. 配体分子的预处理
进行如下操作:
1)点击 Ligand → Input → Open ... 在弹出的对话框中将文件类型由 PDBQT 改为mol2(pdb 格式的配体文件可能会在下一步出现问题),选择“ligand.mol2”,打开我们自己构建的抑制剂配体分子,为配体分子加上极性氢,加上电荷(方法同3.1.1)。
2)点击 Ligand → Torsion Tree → Detect Root… ADT 自动判定 Ligand 的 Root
3)点击 Ligand → Torsion Tree → Show Root Expansion 显示 Root 扩展信息
4)点击 Ligand → Torsion Tree → Show/Hide Root Marker 显示/隐藏 Root 标记
5)点击 Ligand → Torsion Tree → Choose Torsions… 选择 Ligand 中可扭转的键,弹出
6)Torsion Count 对话框中点击 Make all active bonds non-rotatable,Make rotatable bonds between selected atoms rotatable 以及 Make amide bonds rotatable。最后结果如下图所示:该 Ligand 分子 11 个键被设置成可扭转的键(rotatable,绿色),点击 Done 确定并关闭此对话框(注意:通常情况下,一般将环与环之间的单键设置为可旋转的)
7)点击 Ligand → Torsion Tree → Set Number of Torsions… 弹出 Set Number of Active Torsions 对话框,以设置可活动的键的数量,同时指定设定活动键时是需要移动最少的原子(fewest atoms)还是最多的原子(most atoms)。在这里我们设置 fewest atoms,数量为 11,点击 Dismiss 关闭对话框。
8)点击 Ligand → Output → Save as PDBQT… 将准备好的 Ligand 分子保存为“ligand.pdbqt”文件。
需要说明的是:OpenBabel可以快速将分子转化成pdbqt后缀的格式
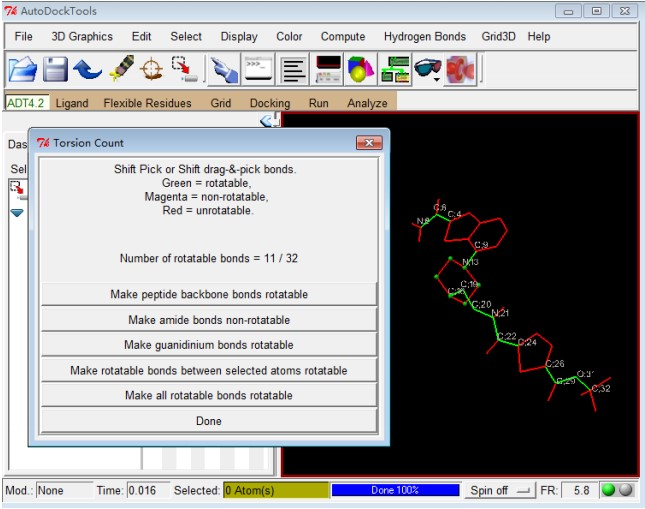
Figure 2: 第六步中Torsion Count 对话框及 Ligand 分子 Torsion 设置效果(绿色圆球表示 Root,绿色键表示可扭转,紫色表示非扭转,红色表示不可扭转)
3.2. Autodock Vina 对接
Vina 是一个命令行程序,它可以和 Autodock Tools 配合使用,因为 Autodock Tools 提供了一个 GUI 界面接口,但是同时,我们也可以在 Windows 的命令提示符程序或 Linux 或是Mac 的终端中执行这个程序。
Note:Vina 是一个相对独立的程序,它的运行可以不依赖 Autodock Tools,也就是说,只要已有了 Vina 所需要的所有输入文件和参数,Vina 就可以进行对接工作。
当然从 Autodock Tools 中调用 Vina 很方便,但是命令行能给予我们更大的发挥空间,特别是后一节将介绍结合使用 Shell 脚本来调用 Vina。
我们这里以 Linux 操作系统为例,假如你使用的是 Ubuntu 或是 Gentoo 等发行版,你可以很方便地使用系统自带的软件包管理程序从镜像上得到并自动安装 Autodock Vina。但是假如你想手动安装 Vina,也是很容易的。方法如下:
从 Vina 的官方下载页面上找到 Linux 的安装包,是一个 tgz 格式的压缩档。使用如下命令解压缩:
~$ tar -xzf autodock_vina_1_1_2_linux_x86.tgz进入解压缩后得到的文件!,进入 bin 子文件!,可以看到 vina "进制可执行文件,为了保#它是可以执行的,我们使用如下命令修改它的执行$限:
~$ chmod +x ./vina如果此时你已经处在 bin 文件!中,使用如下命令就能看到 vina 的版本信息:
~$ ./vina –-help如果你看到了版本信息,就说明你已经安装成功了!
从帮助信息中可以看到,在命令行中我们可以在./vina 之后通过设置参数的方式指定受体、配体文件和格子的信息,但是更加方便的方式是写好一个 configuration 设置脚本,脚本中将需要的参数信息都包(进去,然后)这个脚本直接被 Vina 调用执行。
我们的脚本中需要指定配体和受体的 PDBQT 文件和*+区域的位置和大小,然后调用执行。
~$ ./vina --config /Users/ywtao/Desktop/conf.txt --log /Users/ywtao/Desktop/log.txt#在windows下,我们可以采用下述的代码,首先找到你安装在系统中的vina.exe,然后cmd下运行,
C:\Program Files (x86)\The Scripps Research Institute\Vina>vina --config D:\Data\AutoDock_Screening\HSPB1_1\test\conf.txt --log D:\Data\AutoDock_Screening\HSPB1_1\test\log.txt最后指定的 log 记,文件包(了程序运行时打-在终端窗口中的各种信息。当终端输出时,表明对接工作已经完成。
Writing output ... done.Conf.txt文件的具体内容如下:
receptor = /Users/ywtao/Desktop/rec.pdb
ligand = /Users/ywtao/Desktop/in_17.pdbqt
out = /Users/ywtao/Desktop/all_modes.pdbqt
center_x = 20.031
center_y = 46.672
center_z = -1.889
size_x = 64
size_y = 58
size_z = 503.3. Autodock Vina结果分析
通过查看 log 记,文件,我们可以./ Vina 运行后找到了多少种可能的对接模式,在此例中,我们找到了 9 种。下面介绍如何查看这些对接模式:
1)进入 Autodock Tools,点击 Analyze → Dockings → Open Autodock vina result,打开对接记,文件“all_modes.pdbqt”
2)点击 Analyze → Macromolecule → Open …选择打开受体分子。
3)为受体和配体选择合适的表现形式,通过使用左右方向键切换配体的不同构象,进行观察分析
但是事实上仅仅使用 PyMol 也能进行这样的观察:用 PyMol 打开“all_modes.pdbqt”后,继续打开受体的 pdb 或 pdbqt 文件,同样使用左右方向键观察不同构象的配体与受体的相互作用情况。
- 感谢你赐予我前进的力量
-
 微信
微信  支付宝
支付宝
